Pre-Imputation Quality Control (QC)
Detecting and correcting issues such as genotyping errors, sample handling errors, population stratification etc
is important in GWAS. The preimp_qc module addresses these issues and cleans (QC) your data. Below is a flow diagram
of the filters applied when QC’ing input data:
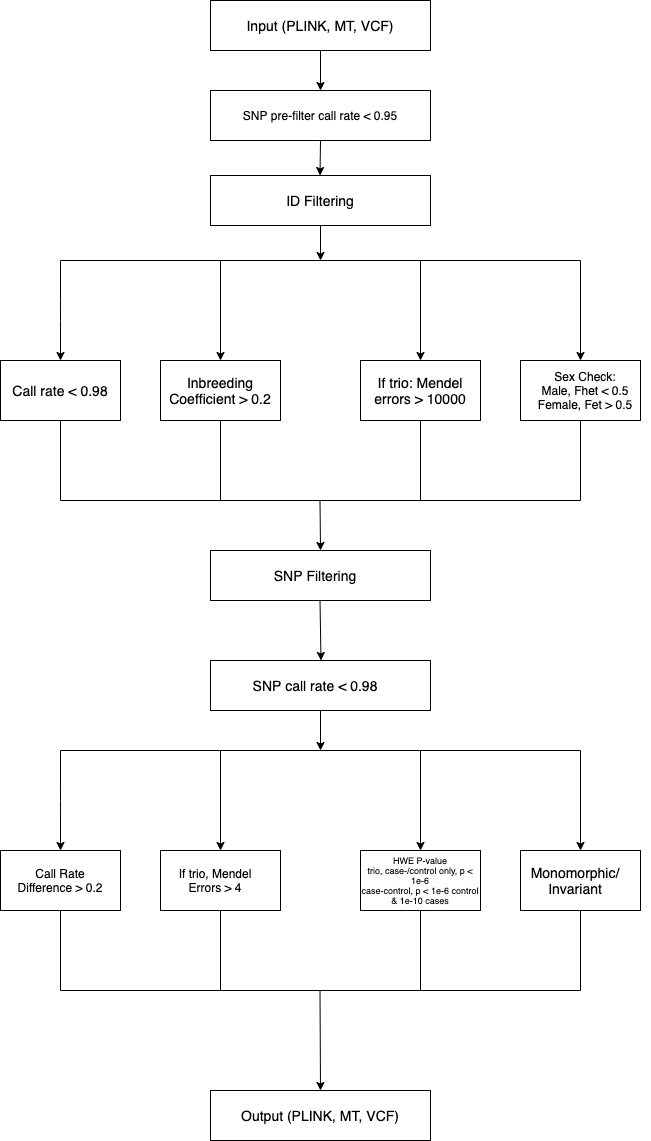
Examples
You can run pre-imputation qc using the preimp_qc module: (1) via the command line; or (2) inside a python script
Command line
preimp_qc --dirname data/ --basename sim_sim2a_eur_sa_merge.miss --input-type plink
Inside a python script
import gwaspy.preimp_qc as qc qc.preimp_qc.preimp_qc(input_type="plink", dirname="data/", basename="sim_sim2a_eur_sa_merge.miss")
Arguments and options
Argument |
Description |
|---|---|
|
Path to where the data is |
|
Data basename |
|
Input type. Options: [ |
|
Export type. Options: [ |
|
Directory path to where output files are going to be saved |
|
Annotations file to be used for annotating sample with information such as Sex and Phenotype |
|
Reference genome build. Default is GRCh38. Options: [ |
|
Generate a QC PDF report or not. Default is True |
|
include only SNPs with missing-rate < NUM (before ID filter), important for post merge of multiple platforms |
|
include only IDs with missing-rate < NUM |
|
include only IDs within NUM < FHET < NUM |
|
include only female IDs with fhet < NUM |
|
include only male IDs with fhet > NUM |
|
include only SNPs with missing-rate < NUM |
|
include only SNPs with missing-rate-difference (case/control) < NUM |
|
include monomorphic (invariant) SNPs |
|
include only SNPs with MAF >= NUM |
|
HWE_controls < NUM |
|
HWE_cases < NUM |
Output(s)
QC’ed file(s) i.e. file with all the variants and/or samples that fail QC filters removed
A detailed PDF QC report including pre- and post-QC variant/sample counts, figures such as Manhattan and QQ plots etc.